原文链接:使用ggplot绘制柱状图,在柱子中显示数值和显著性
本期教程
获得本期教程示例数据,后台回复关键词:20240628。(PS:在社群中,可获得往期和未来教程所有数据和代码)
往期教程部分内容





写在前面
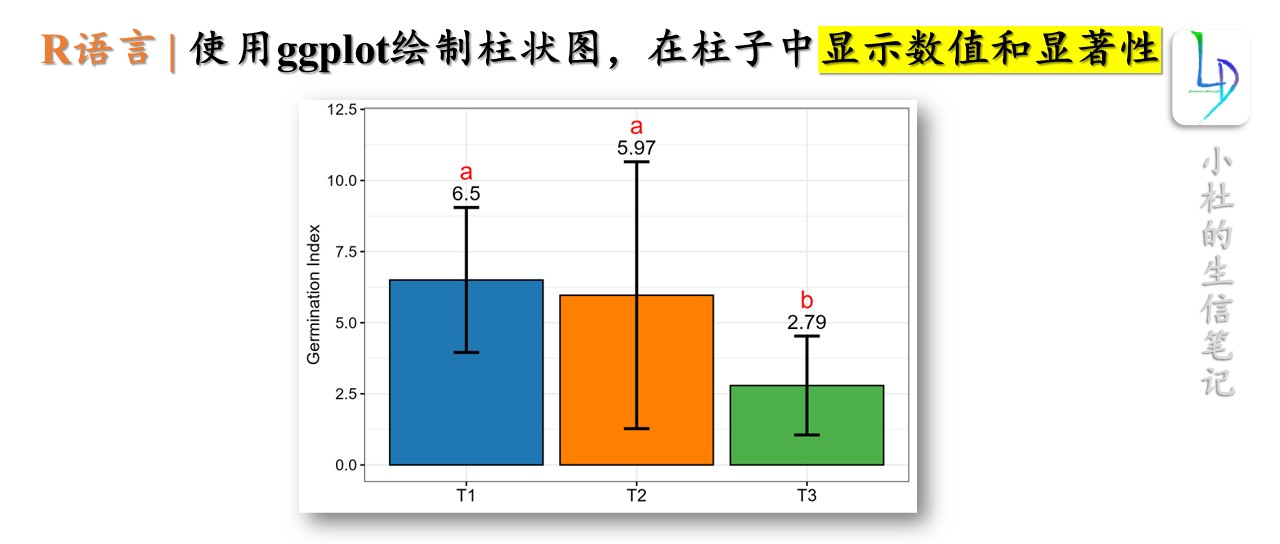
基于ggplot绘制柱状图,小技巧,很基础的图形。但是,越到后面,会发现,越是基础的图形,我们使用的频率越高。今天的教程是基于发芽指数来绘制,我们模拟一个发芽数和天数,计算出发芽指数。
代码
##'@在柱状图中显示数字
##'@2024.06.28
##'@
##'@小杜的生信笔记library(ggplot2)
library(dplyr)
library(tidyr)
library(ggplot2)
library(agricolae)setwd("D:\\BioinfoFile\\小杜的生信笔记\\2024\\20240628_柱状图中显示数据")
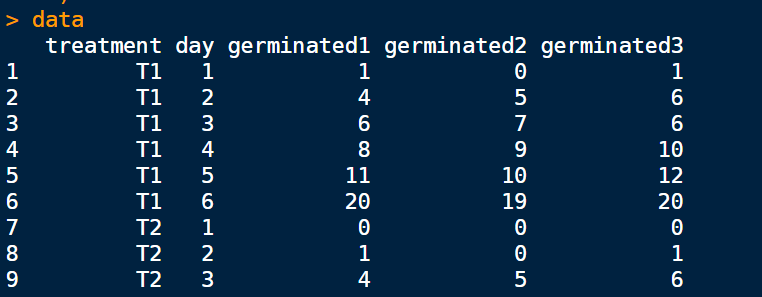
##'@此次数据基于种子发芽指数计算公式进行,数据具有随机性,不具有任何意义。
##'@加载数据
data <- read.csv("20240628_inputdata.csv",header = T)data
相关计算代码
## 转换数据
long_data <- data %>%pivot_longer(cols = starts_with("germinated"), names_to = "replicate", values_to = "germinated")
long_data
##计算活力指数
germination_index <- long_data %>%group_by(treatment, day) %>%summarise(GI = sum(germinated / day)) %>%ungroup()#'@计算各个处理的平均数和标准差
summary_gi <- germination_index %>%group_by(treatment) %>%summarise(mean_GI = mean(GI), sd_GI = sd(GI))#'@单因素方差分析
anova_result <- aov(GI ~ treatment + day, data = germination_index)
summary(anova_result)#'@进行Tukey HSD检验
tukey_result <- HSD.test(anova_result, "treatment", group = TRUE)
print(tukey_result)# 提取 Tukey HSD 结果中的字母标记
groups <- tukey_result$groups
df_letters <- data.frame(sample = rownames(groups), letters = groups$groups)
##'@修改名称
colnames(df_letters) <- c("treatment","letters")# 合并均值和字母标记
df_data <- merge(summary_gi, df_letters, by = "treatment")

绘图
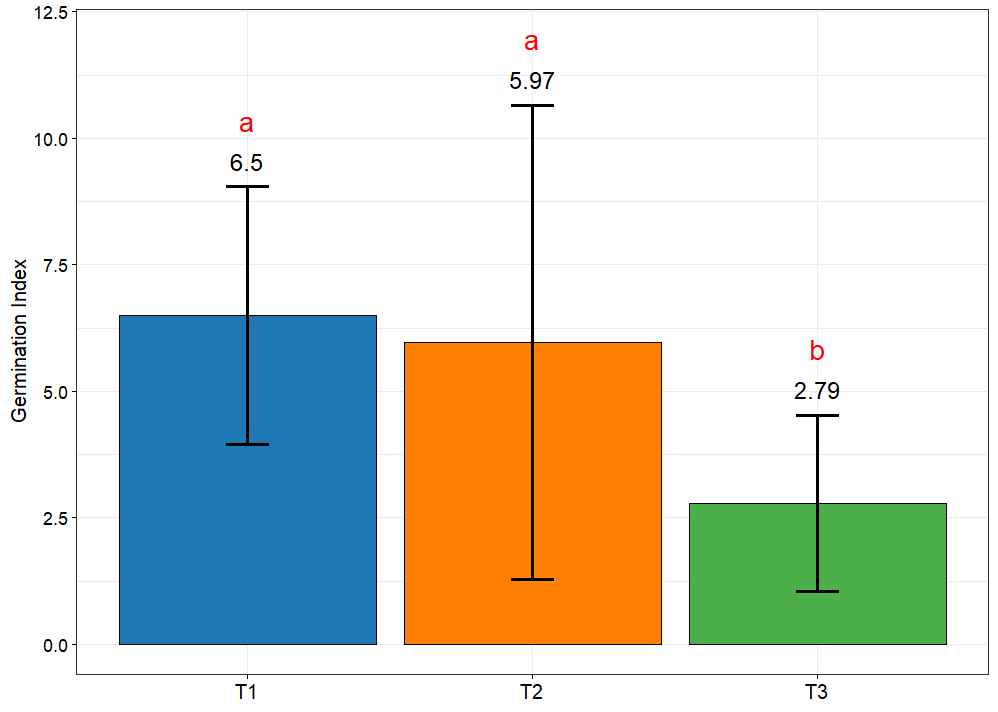
ggplot(df_data, aes(x = treatment, y = mean_GI, fill = treatment)) +geom_bar(stat = "identity", position = "dodge", color = "black", size = 0.5) +##'@误差线geom_errorbar(aes(ymin = mean_GI - sd_GI, ymax = mean_GI + sd_GI), width = 0.15, ## 宽度size = 1 ##字体大小) +##'@显示数字geom_text(aes(x = treatment, y = mean_GI + sd_GI + 0.5, ## “+0.5”显示的高度label = round(mean_GI, 2)), ### "round(mean_GI, 2))",其中2,表示数字显示的小数点位数size = 5,color = 'black') +##'@显示显著性geom_text(aes(x = treatment, y = mean_GI + sd_GI + 1.3, label = letters), size = 6, color = "red") +theme_bw()+scale_fill_manual(values = c("#1f78b4", "#ff7f00", "#4daf4a")) +labs(x = NULL, y = "Germination Index")+theme(#axis.line = element_line(size = 1), ## 粗细text=element_text(#family = "sans",colour ="black",size = 10),axis.text.x = element_text(color = "black", size = 12),axis.text.y = element_text(color = "black",size = 11),axis.ticks = element_line(colour = "black"),strip.text = element_text(color = "black",size = 10),axis.title = element_text(color = "black",size = 12), ##坐标轴字体大小legend.position = "none",strip.background = element_blank())ggsave("20240608_柱状图-显示数字.pdf",width = 6, height = 4)
获得本期教程示例数据,后台回复关键词:20240628。(PS:在社群中,可获得往期和未来教程所有数据和代码)
若我们的教程对你有所帮助,请
点赞+收藏+转发,这是对我们最大的支持。
往期部分文章
1. 最全WGCNA教程(替换数据即可出全部结果与图形)
-
WGCNA分析 | 全流程分析代码 | 代码一
-
WGCNA分析 | 全流程分析代码 | 代码二
-
WGCNA分析 | 全流程代码分享 | 代码三
-
WGCNA分析 | 全流程分析代码 | 代码四
-
WGCNA分析 | 全流程分析代码 | 代码五(最新版本)
2. 精美图形绘制教程
- 精美图形绘制教程
3. 转录组分析教程
-
转录组上游分析教程[零基础]
-
一个转录组上游分析流程 | Hisat2-Stringtie
4. 转录组下游分析
-
批量做差异分析及图形绘制 | 基于DESeq2差异分析
-
GO和KEGG富集分析
-
单基因GSEA富集分析
-
全基因集GSEA富集分析
小杜的生信筆記 ,主要发表或收录生物信息学教程,以及基于R分析和可视化(包括数据分析,图形绘制等);分享感兴趣的文献和学习资料!!